KOSIN UNIVERSITY COLLEGE OF MEDICINE
KOSIN UNIVERSITY COLLEGE OF MEDICINE
Articles
- Page Path
- HOME > Kosin Med J > Volume 38(2); 2023 > Article
-
Review article
Polycystic liver disease: an overview of clinical manifestations, diagnosis, and treatment -
Joonho Jeong1
 , Hyun Joon Park2
, Hyun Joon Park2 -
Kosin Medical Journal 2023;38(2):75-86.
DOI: https://doi.org/10.7180/kmj.23.128
Published online: June 28, 2023
1Department of Internal Medicine, Ulsan University Hospital, University of Ulsan College of Medicine, Ulsan, Korea
2Department of Internal Medicine, Kosin University College of Medicine, Busan, Korea
- Corresponding Author: Hyun Joon Park, MD Department of Internal Medicine, Kosin University College of Medicine, 262 Gamcheon-ro, Seo-gu, Busan 49267, Korea Tel: +82-51-990-6711 Fax: +82-51-990-5055 E-mail: movie406@naver.com
Copyright © 2023 Kosin University College of Medicine.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 2,224 Views
- 78 Download
- 1 Crossref
Abstract
- Polycystic liver disease (PLD) is a hereditary disease characterized by the presence of 20 or more liver cysts. It is classified into three types: isolated autosomal dominant PLD, PLD with autosomal dominant polycystic kidney disease, and PLD with autosomal recessive polycystic kidney disease. Genetic alterations, ciliary dysfunction of the biliary epithelial cells, and aberrant cell signaling pathways are the main factors contributing to the pathophysiology of PLD; however, other complicated mechanisms are also involved. The Gigot and Schnelldorfer classifications are widely used in clinical practice. Most patients with PLD are asymptomatic; however, a few patients with advanced-stage disease may develop symptoms and complications that impair their quality of life and require treatment. The known treatment options for PLD are somatostatin analogues, aspiration with sclerotherapy, fenestration, hepatic resection, and liver transplantation. Although liver transplantation remains the only curative treatment for PLD, medical therapies are gradually being developed with the increasing knowledge of the disease’s pathophysiology. This review focuses on the clinical manifestations and diagnosis of PLD, as well as treatment strategies, to support clinicians regarding the clinical management of the disease.
- Polycystic liver disease (PLD) is a hereditary disease characterized by the presence of ≥20 liver cysts [1]. It presents in three forms: isolated autosomal dominant PLD (ADPLD), PLD with autosomal dominant polycystic kidney disease (ADPKD), and PLD with autosomal recessive polycystic kidney disease (ARPKD). However, the natural course and prognosis are similar among all the PLD classifications [1,2]. Genetic alterations, ciliary dysfunction of the biliary epithelial cells, and aberrant cell signaling pathways are main factors contributing to the complicated pathogenesis of PLD [3-6]. The severity of liver involvement strongly affects clinical symptoms, prognosis, and treatment strategies. Most patients with PLD are asymptomatic; however, patients in advanced stages may develop symptoms, including abdominal pain, gastrointestinal reflux, dyspnea, cystic infection and rupture, malnutrition, ascites, and variceal bleeding. These symptoms and complications seriously affect the quality of life [7,8]. Although liver transplantation is the only curative treatment for PLD, medical therapies are gradually being developed with the increasing knowledge of the disease’s pathophysiology [9-11]. This review focuses on the clinical manifestations, diagnosis, and treatment strategies for PLD to support clinicians in the clinical management of the disease.
Introduction
- 1. ADPLD
- ADPLD is characterized by the presence of multiple liver cysts without renal involvement [1]. The incidence of ADPLD is reported in approximately 1/100,000 individuals worldwide [3]. Various genes are involved in the development of ADPLD; the most prevalent genes are PRKCSH and SEC63 which may account for 20% to 41% of the patients, followed by ALG8, LRP5, GANAB, and SEC61B (Table 1) [1,12-14]. Recently, PKHD1 gene mutation has been proposed to contribute to the development of ADPLD [15]. However, unlike the case with ADPKD and ARPKD, the mentioned genes account for only 30% to 50% of patients with ADPLD, and other pathogenic genes have not been identified in a large number of patients [13].
- 2. PLD with ADPKD
- ADPKD is the most frequent hereditary kidney disease, with a global incidence of 1/500 to 1/1,000 individuals [3]. PLD is the most common extra-renal manifestation of ADPKD [16]. The PKD1 and PKD2 genes have been reported as contributors in the development of ADPKD; PKD1 is reported to be associated with approximately 80% of patients with ADPKD, and PKD2 is reported to be associated with approximately 5% to 10% of patients [4,17]. Moreover, GANAB has recently been shown to be associated with ADPKD [17,18].
- 3. PLD with ARPKD
- ARPKD is a relatively rare hereditary disease with a global incidence of 1/20,000 individuals and occurs mainly in children [3]. PKHD1 gene mutations are associated with ADPKD in most patients [19].
Epidemiology and genetics
- Genetic alterations, ciliary dysfunction of the biliary epithelial cells, and aberrant cell signaling pathways are crucial components in the mechanism of cystic development [4-6,20,21]. Although various genetic alterations contribute to the development of PLD, the exact mechanism is still unclear. To date, the most reliable hypothesis has been the two-hit theory. In addition to the occurrence of a germline mutation (first hit), a somatic mutation (second hit) is required for hepatic cystogenesis [22].
- Most proteins involved in cystogenesis are located in the endoplasmic reticulum (ER). Mutations in the PRKCSH, SEC63, ALG8, LRP5, GANAB, and SEC61B genes are associated with the ER protein glycosylation [12,23]. PRKCSH encodes beta-subunit of glucosidase II and ALG8 encodes a glycosyltransferase, respectively. LRP5 is associated with signaling pathway including Wnt in the cyst development. GANAB encodes the catalytic alpha-subunit of glucosidase II which interacts with the beta-subunit of glucosidase II [24]. In addition, PKD1 and PKD2 genes in ADPKD encode polycystin-1 (PC-1) which is a ciliary protein necessary for cystogenesis, and polycystin-2 (PC-2) which modulates the intracellular calcium levels, respectively. PC-1 and PC-2 form a complex, which works on the surface of the ciliary membrane to simulate calcium uptake [25,26]. Dysfunction of ER proteins inhibits the synthesis, translocation, and expression of other proteins, including PC-1 [25,27]. Therefore, the deficiency in PC-1 expression and the dysfunction of PC-1/PC-2 complexes decrease the intracellular calcium levels and increase the cyclic adenosine monophosphate (cAMP) levels, thus contributing to biliary cell proliferation, fluid secretion, and cystic development [12,21,28].
Pathophysiology
- Most patients with PLD show no clinical symptoms; however, a few patients develop symptoms that decrease their quality of life. The size, number, and location of cysts and the volume of the liver contribute to the development of a range of symptoms, including abdominal pain, abdominal distension, dyspnea, early satiety, gastroesophageal reflux, malnutrition, and back or flank pain caused by hepatomegaly [7-9,29]. The compression of the hepatic veins, portal veins, or inferior vena cava by hepatic cysts can result in a hepatic venous outflow obstruction and portal hypertension, with symptoms such as ascites, variceal bleeding, and splenomegaly [30,31]. Surprisingly, liver failure caused by PLD has been rarely reported; however, it may occur in very severe disease stages [7,14].
- Risk factors associated with the progression of PLD include female sex, older age, multiple pregnancies, and prolonged estrogen exposure [32,33]. Moreover, a cohort study with a large sample size revealed that these factors contribute to symptom development in patients with PLD with ADPKD [14]. Another study revealed that most patients with PLD who underwent liver transplantation were females, suggesting that the disease advances more rapidly in females [34]. The number and size of hepatic cysts in patients with ADPKD are significantly correlated with the number of pregnancies, and some studies have reported an association between estrogen levels and liver volume in patients with PLD [32,33]. This may be supported by the fact that liver volume increases during the reproductive years and then stabilizes after menopause due to decreasing endogenous estrogen [9].
Clinical presentation
- 1. Hepatic cyst infection (HCI)
- HCI is an uncommon complication with an incidence of approximately 1% of patients with hepatic cysts [35]. However, it is an important manifestation of PLD because it may lead to sepsis and death if left untreated. HCI is believed to occur because of the translocation of intestinal bacteremia. Its symptoms include upper right quadrant pain, malaise, and fever [1,36]. Imaging modalities such as computed tomography (CT) scans reveal cystic wall thickening with or without cell debris; however, these modalities are unreliable [36,37]. Recent studies have reported that 18-fluorodeoxyglucose positron emission tomography can be used to confirm HCI by detecting an 18-fluorodeoxyglucose accumulation in the infected cystic epithelia [38]. The gold standard for diagnosing HCI is the identification of inflammatory cells and bacteria via cystic aspiration [1,36,37]. Escherichia coli and Klebsiella spp. are the most prevalent bacteria detected through cystic aspiration [39]. Clinical, biochemical, and radiological parameters can be assessed if cystic aspiration is not possible. Broad-spectrum antibiotic therapy is recommended as the first-line treatment; however, patients with HCI who received only this therapy failed to achieve complete remission. Therefore, cystic drainage combined with antibiotic therapy should be considered [37].
- 2. Hepatic cyst hemorrhage
- Hepatic cyst hemorrhage mainly occurs in large cysts and causes sudden pain in the upper right quadrant or flank [40]. High intra-cystic pressure, rapid cystic growth, and direct trauma are considered triggering factors for cystic hemorrhage. Cystic hemorrhage is diagnosed based on elevated Hounsfield units (due to fibrin aggregation) detected on imaging scans [41,42]. Conservative therapy is recommended in patients with minor symptoms; however, fenestration or hemorrhagic cyst removal can be considered in patients with severe symptoms [40].
- 3. Hepatic cyst rupture
- Cystic rupture is a rare complication of PLD that occurs because of a significant increase in the cystic volume, either spontaneously or after the occurrence of cystic hemorrhage. An increased cystic size may cause instability and increase the risk of rupture. Severe acute abdominal pain is a typical symptom of hepatic cyst rupture. Imaging scans have revealed free fluid around a liver with a residual hepatic cyst. Conservative therapy is recommended in most cases. However, if hemodynamic instability or percutaneous ascites are detected, hepatic cyst drainage or surgical intervention may be required [43,44].
- 4. Portal hypertension
- Patients with severe PLD may experience symptoms such as ascites, variceal hemorrhage, and splenomegaly due to portal hypertension [30]. Despite insufficient data on incidence of portal hypertension in patients with PLD, a retrospective study found that 6% of patients developed portal hypertension during the follow-up period [14]. Portal hypertension due to PLD can be classified into three types: hepatic venous outflow obstruction, portal vein obstruction and/or inferior caval vein syndrome. Most prevalent type is hepatic venous outflow obstruction [30]. Diagnosis is made based on imaging modalities including ultrasound, CT, and magnetic resonance imaging (MRI) by confirming hepatic cysts and ascites [45]. The detection of hepatic venous pressure gradient is needed for diagnosis basically based on the definition of portal hypertension, but it is rarely performed in patients with PLD because it is difficult to conduct due to complicated anatomy and it is usually applied as an assessment tool in patients with cirrhosis. The management of portal hypertension in patients with PLD is still challenging. Diuretics and repeated paracentesis are commonly used for management of ascites. Although there is a lack of data on the effect of somatostatin analogues on ascites control, it can be also used to reduce liver volume. In addition, surgical and interventional approaches for reducing liver volume, transjugular intrahepatic portosystemic shunts, hepatic or portal vein stenting can be considered. Liver transplantation should be considered in patients with refractory ascites or variceal bleeding that is not managed by conventional treatment [30].
Complications
- PLD is usually diagnosed based on the presence of ≥20 hepatic cysts [1]. Recently, the European Association for the Study of the Liver (EASL) has defined PLD as the presence of ≥10 hepatic parenchymal cysts not connected to the bile duct system [9]. A family history of ADPLD, ADPKD, or ARPKD as well as genetic tests can help in establishing the diagnosis. However, only 30% to 50% of patients with ADPLD showed genetic mutations, so genetic tests are not routinely performed to diagnose PLD [9,13,18].
- Liver function tests are usually preserved in for most patients with PLD, because their liver parenchyma is not significantly damaged. Moreover, the carbohydrate antigen 19-9 levels may increase when hepatic cysts occupy an extensive part of the liver parenchyma or when an HCI develops [10,11]. Imaging scans are essential to identify hepatic cysts and exclude other diseases; ultrasonography (USG) and CT are the most widely used diagnostic methods because of their image quality, accessibility, and cost-effectiveness [46]. However, MRI is not routinely performed for diagnosis although it has a high sensitivity for detecting hepatic cysts and can be used to decide suitable treatment options [2,9].
- Currently, three clinical classifications of PLD have been suggested: the Gigot [47], Schnelldorfer[48], and Qian classifications [7]. The Gigot and Schnelldorfer classification systems are widely used in clinical practice. According to the Gigot classification, PLD is classified into three types based on the number and size of hepatic cysts as well as the proportion of the remaining non-cystic liver parenchyma (Table 2). This classification focuses on performing hepatic cyst fenestration in suitable patients with PLD [47]. The Schnelldorfer classification classifies PLD into four types based on symptoms, cyst characteristics, areas of normal liver parenchyma, and portal or hepatic vein occlusion in the preserved sector (Table 3). This classification can be used to decide the optimal treatment [48]. The Qian classification is based on the number of cysts and presence of symptomatic hepatomegaly (Table 4) [7]. However, it is rarely used in clinical practice as it is insufficient for deciding the suitable treatment.
Diagnosis
- Treatment is not required for most asymptomatic patients with PLD; however, it is necessary for the symptomatic minority who have a low quality of life owing to the increased liver volume or complications resulting from PLD [29,49]. The primary goal of PLD treatment is to relieve symptoms and improve the quality of life [9,49]. Several studies have shown that estrogen contributes to the progression of PLD. Consequently, the EASL guidelines discourage female patients with PLD from using oral contraceptives [9].
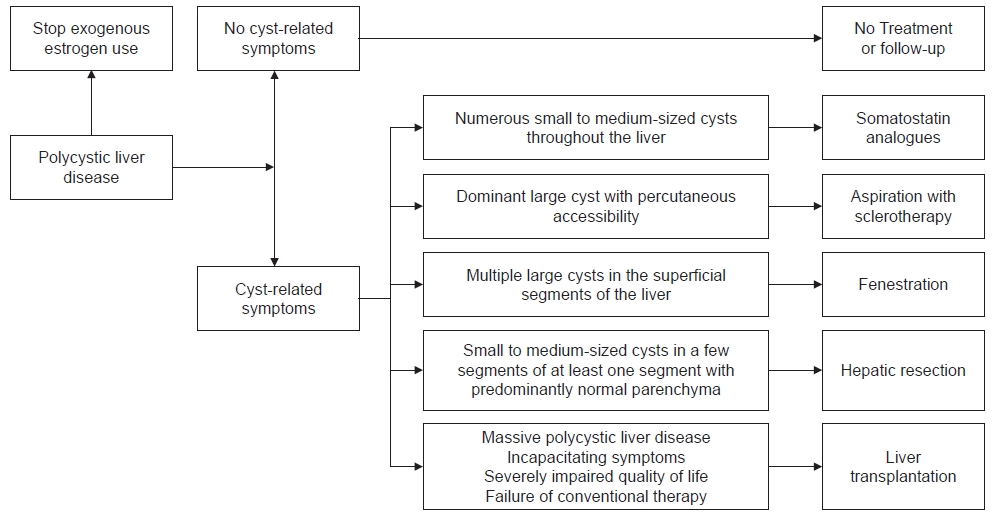
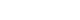
- PLD treatments are classified into three groups: medical, interventional, and surgical. The algorithm for the treatment of PLD is described (Fig. 1) [10]. The optimal treatment is decided according to the number, size, and location of hepatic cysts [9,10].
- 1. Medical treatment
- Somatostatin is a cyclic peptide produced in various tissues, including the gastrointestinal tract and pancreas tissues. It regulates a wide range of physiological functions and hormones by binding to the somatostatin receptors (SSTR), which are classified into five subtypes: SSTR-1 to SSTR-5. The binding of somatostatin analogues to SSTR inhibits cAMP release in cystic cholangiocytes, thus decreasing cystic fluid production and inhibiting bile duct cell hyperplasia, leading to the prevention of hepatic cyst proliferation [50,51].
- Multiple clinical trials have revealed that somatostatin analogues, such as octreotide, lanreotide, and pasireotide, prevent the progression of PLD by decreasing liver volume. Regarding octreotide, Pisani et al. [52] found that the total liver volume was significantly decreased by 130.2±133.2 mL (7.8%±7.4%) in patients who received octreotide per month; however, it was increased by 144.3±316.8 mL (6.1%±14.1%) in patients who received placebo after 3 years of treatment (p=0.004). The total liver volume reduction lasted for 2 years after the treatment completion. Moreover, Hogan et al. [53] reported that the total liver volume was significantly decreased by 4.95%±6.77% in the octreotide group (every 28 days) compared with that in the placebo group after 1 year of treatment (p=0.048). Regarding lanreotide, van Keimpema et al. [54] revealed that the mean liver volume was decreased by 2.9% in patients who received lanreotide per month; however, it was increased by 1.6% in those who received placebo after 6 months of treatment (p=0.01). Pasireotide, a somatostatin analogue with a long half-life, is used to treat Cushing’s syndrome [55]. An animal study revealed that pasireotide is more effective than octreotide in reducing hepatorenal cystogenesis in a rodent model [56]. Similarly, an in vivo study on pasireotide revealed that the total liver volume was changed by –3%±7% in the pasireotide group (every 28 days) compared with the 6%±7% increase in the placebo group. Therefore, researchers concluded that pasireotide slows the progressive-liver-volume increase [57].
- Several meta-analyses have confirmed that somatostatin analogues lower liver volume and improve the quality of life [58-60]. Based on these findings, the EASL guidelines recommend administering somatostatin analogues for patients with numerous small to medium-sized hepatic cysts. The dosage of somatostatin analogues is needed to be adjusted based on efficacy and side effects [9]. In addition, somatostatin analogues are the most effective in young women whose hepatic cysts grow rapidly [61]. Somatostatin analogues are generally considered safe, and the occurrence of serious adverse effects resulting due to cessation is uncommon. Some patients may develop gastrointestinal symptoms (i.e., abdominal discomfort and diarrhea) as well as gallbladder stones [62,63]. Further studies are required to evaluate the effects of long-term maintenance therapy because most studies on somatostatin analogues are based on relatively short treatment periods ranging from 6 months to 3 years.
- mTOR inhibitors are a class of drugs that inhibit mTOR, a serine/threonine-specific protein kinase that belongs to the phosphatidylinositol-3 kinase related kinases family. mTOR regulates various cellular metabolic pathways by signaling through two protein complexes, mTORC1 and mTORC2 [64]. Currently, mTOR inhibitors, including sirolimus and everolimus, are used to treat cancer [65]. In some animal studies on PKD, mTOR inhibitors have been reported to be effective in preventing the growth of hepatic cysts [66]. However, only a few clinical trials on mTOR inhibitors have been conducted, and these mentioned findings have not been confirmed in the clinical trials. Chrispijn et al. [67] evaluated the effectiveness of everolimus plus octreotide versus octreotide monotherapy in reducing liver volume in patients with PLD. No statistically significant differences were detected between the two groups (p=0.73). Furthermore, mTOR inhibitors cause many adverse events including interstitial lung disease, thrombosis, rash, anemia, hyperglycemia, dyslipidemia, and an increased risk of infection [68]. Therefore, mTOR inhibitors are not recommended for the treatment of PLD because of their toxicity and because of insufficient evidence [9].
- V2R is predominantly located in the distal tubules and collecting ducts of the kidney, and its activation induces an elevation in the level of cAMP, which stimulates cell proliferation and hepatic cyst growth [69]. Although V2R antagonists have been clinically confirmed for the treatment of ADPKD [70,71], very few studies have investigated their effectiveness in treating PLD. Only a few case reports have shown that tolvaptan has a favorable effect on reducing liver volume in patients with PLD with ADPKD [72,73]. Therefore, further studies are required to assess the efficacy of V2R antagonists in managing PLD.
- Ursodeoxycholic acid (UDCA), which is widely administered in patients with chronic liver disease, increases intracellular calcium levels in hepatocytes and biliary epithelial cells [74]. Based on this mechanism, it had a favorable effect on delaying the development of hepatic cysts in a rat model. However, only few studies on the efficacy of UDCA in patients with PLD have been conducted. In a phase 2 multicenter randomized controlled study including 34 patients with PLD, total liver volume increased by 4.6%±7.7% after 24 weeks of UDCA treatment compared to 3.1%±3.8% in the control group (p=0.493) [75]. UDCA is not currently recommended for the treatment of PLD according to EASL guidelines [9].
- 2. Interventional treatment
- Cystic aspiration with sclerotherapy is commonly performed for symptomatic patients with dominant large cysts classified as Gigot type I or Schnelldorfer type B to reduce liver volume [1,4,76]. This procedure is conducted under imaging guidance such as USG. After the aspiration of cystic fluid, a sclerosing agent is injected. Ethanol is the most commonly used sclerosing agent, followed by ethanolamine oleate, minocycline, and tetracycline [76,77]. Sclerosing agents destroy the epithelial lining of the cystic wall, thus preventing fluid accumulation within the cyst [78]. Although a single procedure is typically sufficient to treat dominant cysts, some patients require a series of procedures for cystic elimination or to achieve symptomatic relief [79].
- In a systematic analysis of 16 studies assessing the efficacy of aspiration with sclerotherapy for hepatic cysts, including 526 patients with a total of 588 cysts, 76% to 100% of patients reported a partial reduction in the cystic volume, and 72% to 100% reported symptom improvement [80]. Its side effects include postprocedural pain caused by the sclerosing agent and hepatic cyst hemorrhage; however, no fatal complications have been reported [80]. Despite its efficacy for hepatic cysts, aspiration with sclerotherapy is rarely performed in clinical practice because most patients with PLD have multiple hepatic cysts and/or because the cystic size is not suitable for performing the procedure [11].
- TAE for patients with PLD is based on the concept that the cysts are mostly supplied by the hepatic arteries. Impaired blood supply induces the destruction of the cystic epithelial cells, thus reducing cystic fluid accumulation and inhibiting disease progression [81,82]. In a retrospective study on 244 patients with PLD, who underwent TAE, liver volume decreased by 94.7% of the pre-treatment volume within 6 months and by 90.8% within 12 months [83]. However, in a study on 18 patients who underwent TAE, 69.6% experienced TAE failure which included the occurrence of refractory symptoms, cystic infection, liver failure, and death [84]. Therefore, further controlled studies are required to evaluate the efficacy and safety of TAE and to clinically examine this procedure in patients with PLD.
- 3. Surgical treatment
- Cystic fenestration is a surgical procedure that combines aspiration and deroofing of hepatic cysts [85]. Fenestration is usually indicated for patients with Gigot type I-II or Schnelldorfer type B PLD, especially those with cysts in the superficial segments of the liver [6,11]. Additionally, it can be considered when cystic aspiration with sclerotherapy fails. The main advantage of fenestration is that multiple cysts can be treated in a single session [86,87]. Its common complications include ascites, hemorrhage, pleural effusion, and bile leakage [85]. Currently, laparoscopic fenestration is preferred to open fenestration as it has the advantages of shorter hospital-stay duration, fewer complications, and the absence of significant differences regarding the outcomes. Nevertheless, open fenestration is appropriate in cysts located in difficult-to-access regions such as the hepatic dome or right posterior segments [85,87]. In a meta-analysis that assessed symptomatic relief and recurrence after laparoscopic fenestration in 1,314 patients with hepatic cysts, 90.2% of the patients showed symptomatic relief. The rates of symptomatic recurrence and re-intervention were 9.6% and 7.1%, respectively. However, in a subgroup analysis on patients with PLD (n=146), the rates of symptomatic recurrence, re-intervention, and complications were significantly higher (33.7%, 26.4%, and 29.3%, respectively) [88]. Cystic fenestration is considered a suitable treatment option in patients with symptomatic large cysts.
- Hepatic resection is primarily performed in symptomatic Gigot type II or Schnelldorfer type C PLD with small to medium-sized cysts in a few segments with at least one segment having predominantly normal liver parenchyma [11,48]. In addition, it can be considered when aspiration with sclerotherapy, cystic fenestration, or liver transplantation is not available [6,10]. Hepatic resection results in a significant reduction in liver volume and symptomatic relief. However, the biliary tract, vascular structure, and Glisson’s capsule distortions caused by cystic compression and hepatic venous outflow obstruction complicate the operation and increase morbidity and mortality rates. The main complications include intra- or post-operative hemorrhage, ascites, pleural effusion, and liver failure [4,86]. A review of 26 articles on 337 patients with PLD who underwent hepatectomy revealed that 86% of the patients showed symptomatic relief. The morbidity and mortality rates were 51% and 3%, respectively. Cystic recurrence was noted in 34% of the patients [86]. Currently, studies on combining partial hepatectomy with cystic fenestration [85] and somatostatin analogues administration after hepatectomy in patients with PLD [89] have shown favorable outcomes in terms of liver volume reduction and hepatic cyst growth suppression. Although hepatic resection relieves symptoms and reduces liver volume, we should keep in mind its considerable morbidity and mortality rates, as well as the potential challenges we may encounter when performing future liver transplantation due to the occurrence of adhesions [4,10,86,90].
- Liver transplantation is the only curative treatment for PLD and is mostly performed in patients with Gigot type III or Schnelldorfer type D disease [4,9,10,48]. Patients with incapacitating symptoms such as severe malnutrition, portal hypertension, ascites, variceal bleeding, or recurrent HCIs or those who have failed conventional therapy should be considered for undergoing liver transplantation [91,92]. According to the European Liver Transplant Registry study, the 1- and 5-year graft survival rates were 94.3% and 87.5%, and the 1- and 5-year survival rates were 94.8% and 92.3%, respectively [34].
- The Model for End-Stage Liver Disease (MELD) score, which has been validated in patients with liver cirrhosis, is currently the main criteria for deciding to allocate liver grafts [93,94]. However, it is difficult to allocate liver grafts in patients with PLD because of the low MELD scores, as liver function remains preserved even in the most advanced stage. As a result, several exceptions have been proposed to take patients with PLD on waiting lists into consideration. support patients with PLD on waiting lists [91,95]. These guidelines need to be applied more effectively in the allocation system, and more beneficial criteria are required.
- Combined liver-kidney transplantation should be considered in patients with PLD with ADPKD and severe renal impairment (creatinine clearance <30 mL/min) since the outcome of this combined technique has been better than that of liver transplantation alone [96,97]. However, combined liver-kidney transplantation have been raised rates of short-term kidney graft loss about 20% [98]. Therefore, a multidisciplinary approach for decision-making in selecting patients, involving hepatologists and nephrologists, is recommended.
Treatment
1) Somatostatin analogues
2) Mechanistic target of rapamycin (mTOR) inhibitors
3) Vasopressin-2 receptor (V2R) antagonists
4) Ursodeoxycholic acid
1) Cystic aspiration with sclerotherapy
2) Transcatheter arterial embolization (TAE)
1) Fenestration
2) Hepatic resection
3) Liver transplantation
- PLD is an inherited genetic disorder. Only a few patients develop symptoms and complications that require further treatment. Its diagnosis is based on a family history of the disease and the presence of multiple hepatic cysts, with or without renal cysts, via imaging modalities. The primary goal of PLD treatment is to relieve symptoms and improve the patients’ quality of life. Liver transplantation is the only curative treatment option; however, it is not available for many patients. In addition to liver transplantation, medical therapies including somatostatin analogues, aspiration with sclerotherapy, TAE, fenestration, and liver resection can be carefully applied in certain situations. Future effective pharmacological treatments or combination therapies may be developed based on a better understanding of the PLD pathophysiology.
Conclusion
-
Conflicts of interest
Hyun Joon Park is an editorial board member of the journal but was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
-
Funding
None.
-
Author contributions
Conceptualization: HJP. Supervision: HJP. Writing – original draft: JJ. Writing – review & editing: HJP.
Article information
| Disease | Gene |
|---|---|
| ADPLD | PRKCSH, SEC63, ALG8, LRP5, GANAB, SEC61B, PKHD1 |
| ADPKD | PKD1, PKD2, GANAB |
| ARPKD | PKHD1 |
| Qian classification | No. of cysts | Symptomatic hepatomegaly |
|---|---|---|
| Grade 0 | 0 | Absent |
| Grade 1 | 1–10 | Absent |
| Grade 2 | 11–20 | Absent |
| Grade 3 | >20 | Absent |
| Grade 4 | >20 | Present |
- 1. Gevers TJ, Drenth JP. Diagnosis and management of polycystic liver disease. Nat Rev Gastroenterol Hepatol 2013;10:101–8.ArticlePubMedPDF
- 2. Chandok N. Polycystic liver disease: a clinical review. Ann Hepatol 2012;11:819–26.ArticlePubMed
- 3. Perugorria MJ, Masyuk TV, Marin JJ, Marzioni M, Bujanda L, LaRusso NF, et al. Polycystic liver diseases: advanced insights into the molecular mechanisms. Nat Rev Gastroenterol Hepatol 2014;11:750–61.ArticlePubMedPMCPDF
- 4. Wong MY, McCaughan GW, Strasser SI. An update on the pathophysiology and management of polycystic liver disease. Expert Rev Gastroenterol Hepatol 2017;11:569–81.ArticlePubMed
- 5. Santos-Laso A, Izquierdo-Sanchez L, Rodrigues PM, Huang BQ, Azkargorta M, Lapitz A, et al. Proteostasis disturbances and endoplasmic reticulum stress contribute to polycystic liver disease: new therapeutic targets. Liver Int 2020;40:1670–85.ArticlePubMedPMCPDF
- 6. Norcia LF, Watanabe EM, Hamamoto Filho PT, Hasimoto CN, Pelafsky L, de Oliveira WK, et al. Polycystic liver disease: pathophysiology, diagnosis and treatment. Hepat Med 2022;14:135–61.ArticlePubMedPMCPDF
- 7. Qian Q, Li A, King BF, Kamath PS, Lager DJ, Huston J 3rd, et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology 2003;37:164–71.ArticlePubMed
- 8. Bistritz L, Tamboli C, Bigam D, Bain VG. Polycystic liver disease: experience at a teaching hospital. Am J Gastroenterol 2005;100:2212–7.ArticlePubMed
- 9. European Association for the Study of the Liver. EASL Clinical Practice Guidelines on the management of cystic liver diseases. J Hepatol 2022;77:1083–108.ArticlePubMed
- 10. van Aerts RMM, van de Laarschot LFM, Banales JM, Drenth JPH. Clinical management of polycystic liver disease. J Hepatol 2018;68:827–37.ArticlePubMed
- 11. Zhang ZY, Wang ZM, Huang Y. Polycystic liver disease: classification, diagnosis, treatment process, and clinical management. World J Hepatol 2020;12:72–83.ArticlePubMedPMC
- 12. Besse W, Dong K, Choi J, Punia S, Fedeles SV, Choi M, et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J Clin Invest 2017;127:1772–85.ArticlePubMedPMC
- 13. Besse W, Choi J, Ahram D, Mane S, Sanna-Cherchi S, Torres V, et al. A noncoding variant in GANAB explains isolated polycystic liver disease (PCLD) in a large family. Hum Mutat 2018;39:378–82.ArticlePubMedPMCPDF
- 14. Van Keimpema L, De Koning DB, Van Hoek B, Van Den Berg AP, Van Oijen MG, De Man RA, et al. Patients with isolated polycystic liver disease referred to liver centres: clinical characterization of 137 cases. Liver Int 2011;31:92–8.ArticlePubMed
- 15. Lanktree MB, Haghighi A, Guiard E, Iliuta IA, Song X, Harris PC, et al. Prevalence estimates of polycystic kidney and liver disease by population sequencing. J Am Soc Nephrol 2018;29:2593–600.ArticlePubMedPMC
- 16. Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 2010;17:173–80.ArticlePubMed
- 17. Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers 2018;4:50.ArticlePubMedPMCPDF
- 18. Cornec-Le Gall E, Torres VE, Harris PC. Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J Am Soc Nephrol 2018;29:13–23.ArticlePubMedPMC
- 19. Bergmann C, Senderek J, Kupper F, Schneider F, Dornia C, Windelen E, et al. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat 2004;23:453–63.ArticlePubMed
- 20. Fabris L, Fiorotto R, Spirli C, Cadamuro M, Mariotti V, Perugorria MJ, et al. Pathobiology of inherited biliary diseases: a roadmap to understand acquired liver diseases. Nat Rev Gastroenterol Hepatol 2019;16:497–511.ArticlePubMedPMCPDF
- 21. Kim S, Nie H, Nesin V, Tran U, Outeda P, Bai CX, et al. The polycystin complex mediates Wnt/Ca(2+) signalling. Nat Cell Biol 2016;18:752–64.ArticlePubMedPMCPDF
- 22. Janssen MJ, Waanders E, Te Morsche RH, Xing R, Dijkman HB, Woudenberg J, et al. Secondary, somatic mutations might promote cyst formation in patients with autosomal dominant polycystic liver disease. Gastroenterology 2011;141:2056–63.ArticlePubMed
- 23. Breitling J, Aebi M. N-linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harb Perspect Biol 2013;5:a013359.ArticlePubMedPMC
- 24. Yu Z, Shen X, Hu C, Zeng J, Wang A, Chen J. Molecular mechanisms of isolated polycystic liver diseases. Front Genet 2022;13:846877.ArticlePubMedPMC
- 25. Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004;279:40419–30.ArticlePubMed
- 26. Ghata J, Cowley BD Jr. Polycystic kidney disease. Compr Physiol 2017;7:945–75.ArticlePubMedPDF
- 27. Fedeles SV, Gallagher AR, Somlo S. Polycystin-1: a master regulator of intersecting cystic pathways. Trends Mol Med 2014;20:251–60.ArticlePubMedPMC
- 28. Su X, Wu M, Yao G, El-Jouni W, Luo C, Tabari A, et al. Regulation of polycystin-1 ciliary trafficking by motifs at its C-terminus and polycystin-2 but not by cleavage at the GPS site. J Cell Sci 2015;128:4063–73.ArticlePubMedPMC
- 29. Neijenhuis MK, Kievit W, Verheesen SM, D’Agnolo HM, Gevers TJ, Drenth JP. Impact of liver volume on polycystic liver disease-related symptoms and quality of life. United European Gastroenterol J 2018;6:81–8.ArticlePubMedPMCPDF
- 30. Bernts LH, Drenth JPH, Tjwa ET. Management of portal hypertension and ascites in polycystic liver disease. Liver Int 2019;39:2024–33.ArticlePubMedPMCPDF
- 31. Bernts LHP, Tjwa ET, D’Agnolo HM, Jenniskens SF, Drenth JP. Venous stent placement for refractory ascites due to hepatic venous outflow obstruction in polycystic liver disease. J Vasc Interv Radiol 2019;30:1617–9.ArticlePubMed
- 32. Gabow PA, Johnson AM, Kaehny WD, Manco-Johnson ML, Duley IT, Everson GT. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology 1990;11:1033–7.ArticlePubMed
- 33. Sherstha R, McKinley C, Russ P, Scherzinger A, Bronner T, Showalter R, et al. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology 1997;26:1282–6.ArticlePubMed
- 34. van Keimpema L, Nevens F, Adam R, Porte RJ, Fikatas P, Becker T, et al. Excellent survival after liver transplantation for isolated polycystic liver disease: an European Liver Transplant Registry study. Transpl Int 2011;24:1239–45.ArticlePubMed
- 35. Chebib FT, Jung Y, Heyer CM, Irazabal MV, Hogan MC, Harris PC, et al. Effect of genotype on the severity and volume progression of polycystic liver disease in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2016;31:952–60.ArticlePubMedPMC
- 36. Suwabe T, Ubara Y, Higa Y, Nakanishi S, Sogawa Y, Nomura K, et al. Infected hepatic and renal cysts: differential impact on outcome in autosomal dominant polycystic kidney disease. Nephron Clin Pract 2009;112:c157–63.ArticlePubMedPDF
- 37. Lantinga MA, Drenth JP, Gevers TJ. Diagnostic criteria in renal and hepatic cyst infection. Nephrol Dial Transplant 2015;30:744–51.ArticlePubMed
- 38. Jouret F, Lhommel R, Beguin C, Devuyst O, Pirson Y, Hassoun Z, et al. Positron-emission computed tomography in cyst infection diagnosis in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2011;6:1644–50.ArticlePubMed
- 39. Suwabe T, Araoka H, Ubara Y, Kikuchi K, Hazue R, Mise K, et al. Cyst infection in autosomal dominant polycystic kidney disease: causative microorganisms and susceptibility to lipid-soluble antibiotics. Eur J Clin Microbiol Infect Dis 2015;34:1369–79.ArticlePubMedPDF
- 40. Fong ZV, Wolf AM, Doria C, Berger AC, Rosato EL, Palazzo F. Hemorrhagic hepatic cyst: report of a case and review of the literature with emphasis on clinical approach and management. J Gastrointest Surg 2012;16:1782–9.ArticlePubMedPDF
- 41. Mortele KJ, Ros PR. Cystic focal liver lesions in the adult: differential CT and MR imaging features. Radiographics 2001;21:895–910.ArticlePubMed
- 42. Everson GT, Scherzinger A, Berger-Leff N, Reichen J, Lezotte D, Manco-Johnson M, et al. Polycystic liver disease: quantitation of parenchymal and cyst volumes from computed tomography images and clinical correlates of hepatic cysts. Hepatology 1988;8:1627–34.ArticlePubMed
- 43. Marion Y, Brevartt C, Plard L, Chiche L. Hemorrhagic liver cyst rupture: an unusual life-threatening complication of hepatic cyst and literature review. Ann Hepatol 2013;12:336–9.ArticlePubMed
- 44. Miliadis L, Giannakopoulos T, Boutsikos G, Terzis I, Kyriazanos ID. Spontaneous rupture of a large non-parasitic liver cyst: a case report. J Med Case Rep 2010;4:2.ArticlePubMedPMCPDF
- 45. Rajoriya N, Tripathi D, Leithead JA, Gunson BK, Lord S, Ferguson JW, et al. Portal hypertension in polycystic liver disease patients does not affect wait-list or immediate post-liver transplantation outcomes. World J Gastroenterol 2016;22:9966–73.ArticlePubMedPMC
- 46. Levine E, Cook LT, Grantham JJ. Liver cysts in autosomal-dominant polycystic kidney disease: clinical and computed tomographic study. AJR Am J Roentgenol 1985;145:229–33.ArticlePubMed
- 47. Gigot JF, Jadoul P, Que F, Van Beers BE, Etienne J, Horsmans Y, et al. Adult polycystic liver disease: is fenestration the most adequate operation for long-term management? Ann Surg 1997;225:286–94.ArticlePubMedPMC
- 48. Schnelldorfer T, Torres VE, Zakaria S, Rosen CB, Nagorney DM. Polycystic liver disease: a critical appraisal of hepatic resection, cyst fenestration, and liver transplantation. Ann Surg 2009;250:112–8.ArticlePubMed
- 49. van Keimpema L, Hockerstedt K. Treatment of polycystic liver disease. Br J Surg 2009;96:1379–80.ArticlePubMedPDF
- 50. Tietz PS, Alpini G, Pham LD, Larusso NF. Somatostatin inhibits secretin-induced ductal hypercholeresis and exocytosis by cholangiocytes. Am J Physiol 1995;269(1 Pt 1):G110–8.Article
- 51. Tan CK, Podila PV, Taylor JE, Nagorney DM, Wiseman GA, Gores GJ, et al. Human cholangiocarcinomas express somatostatin receptors and respond to somatostatin with growth inhibition. Gastroenterology 1995;108:1908–16.ArticlePubMed
- 52. Pisani A, Sabbatini M, Imbriaco M, Riccio E, Rubis N, Prinster A, et al. Long-term effects of octreotide on liver volume in patients with polycystic kidney and liver disease. Clin Gastroenterol Hepatol 2016;14:1022–30.ArticlePubMed
- 53. Hogan MC, Masyuk TV, Page LJ, Kubly VJ, Bergstralh EJ, Li X, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol 2010;21:1052–61.ArticlePubMedPMC
- 54. van Keimpema L, Nevens F, Vanslembrouck R, van Oijen MG, Hoffmann AL, Dekker HM, et al. Lanreotide reduces the volume of polycystic liver: a randomized, double-blind, placebo-controlled trial. Gastroenterology 2009;137:1661–8.ArticlePubMed
- 55. Lesche S, Lehmann D, Nagel F, Schmid HA, Schulz S. Differential effects of octreotide and pasireotide on somatostatin receptor internalization and trafficking in vitro. J Clin Endocrinol Metab 2009;94:654–61.ArticlePubMedPDF
- 56. Masyuk TV, Radtke BN, Stroope AJ, Banales JM, Gradilone SA, Huang B, et al. Pasireotide is more effective than octreotide in reducing hepatorenal cystogenesis in rodents with polycystic kidney and liver diseases. Hepatology 2013;58:409–21.ArticlePubMedPMC
- 57. Hogan MC, Chamberlin JA, Vaughan LE, Waits AL, Banks C, Leistikow K, et al. Pansomatostatin agonist pasireotide long-acting release for patients with autosomal dominant polycystic kidney or liver disease with severe liver involvement: a randomized clinical trial. Clin J Am Soc Nephrol 2020;15:1267–78.ArticlePubMedPMC
- 58. Griffiths J, Mills MT, Ong AC. Long-acting somatostatin analogue treatments in autosomal dominant polycystic kidney disease and polycystic liver disease: a systematic review and meta-analysis. BMJ Open 2020;10:e032620.ArticlePubMedPMC
- 59. Suwabe T, Barrera FJ, Rodriguez-Gutierrez R, Ubara Y, Hogan MC. Somatostatin analog therapy effectiveness on the progression of polycystic kidney and liver disease: a systematic review and meta-analysis of randomized clinical trials. PLoS One 2021;16:e0257606.ArticlePubMedPMC
- 60. Garofalo C, Capuano I, Pennino L, De Gregorio I, Riccio E, Provenzano M, et al. The effects of somatostatin analogues on liver volume and quality of life in polycystic liver disease: a meta-analysis of randomized controlled trials. Sci Rep 2021;11:23500.ArticlePubMedPMCPDF
- 61. Gevers TJ, Inthout J, Caroli A, Ruggenenti P, Hogan MC, Torres VE, et al. Young women with polycystic liver disease respond best to somatostatin analogues: a pooled analysis of individual patient data. Gastroenterology 2013;145:357–65.ArticlePubMed
- 62. Santos-Laso A, Izquierdo-Sanchez L, Lee-Law PY, Perugorria MJ, Marzioni M, Marin JJ, et al. New advances in polycystic liver diseases. Semin Liver Dis 2017;37:45–55.ArticlePubMed
- 63. Temmerman F, Gevers T, Ho TA, Vanslembrouck R, Coudyzer W, van Pelt J, et al. Safety and efficacy of different lanreotide doses in the treatment of polycystic liver disease: pooled analysis of individual patient data. Aliment Pharmacol Ther 2013;38:397–406.ArticlePubMed
- 64. Ren XS, Sato Y, Harada K, Sasaki M, Furubo S, Song JY, et al. Activation of the PI3K/mTOR pathway is involved in cystic proliferation of cholangiocytes of the PCK rat. PLoS One 2014;9:e87660.ArticlePubMedPMC
- 65. Populo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. Int J Mol Sci 2012;13:1886–918.ArticlePubMedPMC
- 66. Temmerman F, Chen F, Libbrecht L, Vander Elst I, Windmolders P, Feng Y, et al. Everolimus halts hepatic cystogenesis in a rodent model of polycystic-liver-disease. World J Gastroenterol 2017;23:5499–507.ArticlePubMedPMC
- 67. Chrispijn M, Gevers TJ, Hol JC, Monshouwer R, Dekker HM, Drenth JP. Everolimus does not further reduce polycystic liver volume when added to long acting octreotide: results from a randomized controlled trial. J Hepatol 2013;59:153–9.ArticlePubMed
- 68. Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf 2013;12:177–86.ArticlePubMed
- 69. Gattone VH 2nd, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003;9:1323–6.ArticlePubMedPDF
- 70. Wang X, Gattone V 2nd, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005;16:846–51.ArticlePubMed
- 71. Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012;367:2407–18.ArticlePubMedPMC
- 72. Takenaka T, Miura S, Kitajima M. The management of polycystic liver disease by tolvaptan. Clin Mol Hepatol 2020;26:70–3.ArticlePubMedPMCPDF
- 73. Mizuno H, Hoshino J, Suwabe T, Sumida K, Sekine A, Oshima Y, et al. Tolvaptan for the treatment of enlarged polycystic liver disease. Case Rep Nephrol Dial 2017;7:108–11.ArticlePubMedPMCPDF
- 74. Munoz-Garrido P, Marin JJ, Perugorria MJ, Urribarri AD, Erice O, Saez E, et al. Ursodeoxycholic acid inhibits hepatic cystogenesis in experimental models of polycystic liver disease. J Hepatol 2015;63:952–61.ArticlePubMedPMC
- 75. D'Agnolo HM, Kievit W, Takkenberg RB, Riano I, Bujanda L, Neijenhuis MK, et al. Ursodeoxycholic acid in advanced polycystic liver disease: a phase 2 multicenter randomized controlled trial. J Hepatol 2016;65:601–7.ArticlePubMed
- 76. Yamada N, Shinzawa H, Ukai K, Makino N, Matsuhashi T, Wakabayashi H, et al. Treatment of symptomatic hepatic cysts by percutaneous instillation of minocycline hydrochloride. Dig Dis Sci 1994;39:2503–9.ArticlePubMedPDF
- 77. Nakaoka R, Das K, Kudo M, Chung H, Innoue T. Percutaneous aspiration and ethanolamine oleate sclerotherapy for sustained resolution of symptomatic polycystic liver disease: an initial experience. AJR Am J Roentgenol 2009;193:1540–5.ArticlePubMed
- 78. van Keimpema L, de Koning DB, Strijk SP, Drenth JP. Aspiration-sclerotherapy results in effective control of liver volume in patients with liver cysts. Dig Dis Sci 2008;53:2251–7.ArticlePubMedPMC
- 79. Kairaluoma MI, Leinonen A, Stahlberg M, Paivansalo M, Kiviniemi H, Siniluoto T. Percutaneous aspiration and alcohol sclerotherapy for symptomatic hepatic cysts: an alternative to surgical intervention. Ann Surg 1989;210:208–15.ArticlePubMedPMC
- 80. Wijnands TF, Gortjes AP, Gevers TJ, Jenniskens SF, Kool LJ, Potthoff A, et al. Efficacy and safety of aspiration sclerotherapy of simple hepatic cysts: a systematic review. AJR Am J Roentgenol 2017;208:201–7.ArticlePubMed
- 81. Ubara Y, Takei R, Hoshino J, Tagami T, Sawa N, Yokota M, et al. Intravascular embolization therapy in a patient with an enlarged polycystic liver. Am J Kidney Dis 2004;43:733–8.ArticlePubMed
- 82. Park HC, Kim CW, Ro H, Moon JY, Oh KH, Kim Y, et al. Transcatheter arterial embolization therapy for a massive polycystic liver in autosomal dominant polycystic kidney disease patients. J Korean Med Sci 2009;24:57–61.ArticlePubMedPMC
- 83. Hoshino J, Ubara Y, Suwabe T, Sumida K, Hayami N, Mise K, et al. Intravascular embolization therapy in patients with enlarged polycystic liver. Am J Kidney Dis 2014;63:937–44.ArticlePubMed
- 84. Yang J, Ryu H, Han M, Kim H, Hwang YH, Chung JW, et al. Comparison of volume-reductive therapies for massive polycystic liver disease in autosomal dominant polycystic kidney disease. Hepatol Res 2016;46:183–91.ArticlePubMedPDF
- 85. Russell RT, Pinson CW. Surgical management of polycystic liver disease. World J Gastroenterol 2007;13:5052–9.ArticlePubMedPMC
- 86. Drenth JP, Chrispijn M, Nagorney DM, Kamath PS, Torres VE. Medical and surgical treatment options for polycystic liver disease. Hepatology 2010;52:2223–30.ArticlePubMed
- 87. van Keimpema L, Ruurda JP, Ernst MF, van Geffen HJ, Drenth JP. Laparoscopic fenestration of liver cysts in polycystic liver disease results in a median volume reduction of 12.5%. J Gastrointest Surg 2008;12:477–82.ArticlePubMedPDF
- 88. Bernts LHP, Echternach SG, Kievit W, Rosman C, Drenth JPH. Clinical response after laparoscopic fenestration of symptomatic hepatic cysts: a systematic review and meta-analysis. Surg Endosc 2019;33:691–704.ArticlePubMedPMCPDF
- 89. Tseng J, Orloff SL. Management of symptomatic polycystic liver disease with hepatic resection. JAMA Surg 2015;150:81–2.ArticlePubMed
- 90. Aussilhou B, Doufle G, Hubert C, Francoz C, Paugam C, Paradis V, et al. Extended liver resection for polycystic liver disease can challenge liver transplantation. Ann Surg 2010;252:735–43.ArticlePubMed
- 91. Arrazola L, Moonka D, Gish RG, Everson GT. Model for end-stage liver disease (MELD) exception for polycystic liver disease. Liver Transpl 2006;12(12 Suppl 3):S110–1.Article
- 92. Aussilhou B, Dokmak S, Dondero F, Joly D, Durand F, Soubrane O, et al. Treatment of polycystic liver disease: update on the management. J Visc Surg 2018;155:471–81.ArticlePubMed
- 93. Wiesner R, Edwards E, Freeman R, Harper A, Kim R, Kamath P, et al. Model for end-stage liver disease (MELD) and allocation of donor livers. Gastroenterology 2003;124:91–6.ArticlePubMed
- 94. Neuberger J, Gimson A, Davies M, Akyol M, O'Grady J, Burroughs A, et al. Selection of patients for liver transplantation and allocation of donated livers in the UK. Gut 2008;57:252–7.ArticlePubMed
- 95. Freeman RB Jr, Gish RG, Harper A, Davis GL, Vierling J, Lieblein L, et al. Model for end-stage liver disease (MELD) exception guidelines: results and recommendations from the MELD Exception Study Group and Conference (MESSAGE) for the approval of patients who need liver transplantation with diseases not considered by the standard MELD formula. Liver Transpl 2006;12(12 Suppl 3):S128–36.Article
- 96. Coquillard C, Berger J, Daily M, Shah M, Mei X, Marti F, et al. Combined liver-kidney transplantation for polycystic liver and kidney disease: analysis from the United Network for Organ Sharing dataset. Liver Int 2016;36:1018–25.ArticlePubMedPDF
- 97. Simpson N, Cho YW, Cicciarelli JC, Selby RR, Fong TL. Comparison of renal allograft outcomes in combined liver-kidney transplantation versus subsequent kidney transplantation in liver transplant recipients: analysis of UNOS Database. Transplantation 2006;82:1298–303.ArticlePubMed
- 98. Lunsford KE, Bodzin AS, Markovic D, Zarrinpar A, Kaldas FM, Gritsch HA, et al. Avoiding futility in simultaneous liver-kidney transplantation: analysis of 331 consecutive patients listed for dual organ replacement. Ann Surg 2017;265:1016–24.ArticlePubMed
References
Figure & Data
References
Citations
- Predicting Safe Liver Resection Volume for Major Hepatectomy Using Artificial Intelligence
Chol Min Kang, Hyung June Ku, Hyung Hwan Moon, Seong-Eun Kim, Ji Hoon Jo, Young Il Choi, Dong Hoon Shin
Journal of Clinical Medicine.2024; 13(2): 381. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite