Polycystic liver disease: an overview of clinical manifestations, diagnosis, and treatment
Article information

Abstract
Polycystic liver disease (PLD) is a hereditary disease characterized by the presence of 20 or more liver cysts. It is classified into three types: isolated autosomal dominant PLD, PLD with autosomal dominant polycystic kidney disease, and PLD with autosomal recessive polycystic kidney disease. Genetic alterations, ciliary dysfunction of the biliary epithelial cells, and aberrant cell signaling pathways are the main factors contributing to the pathophysiology of PLD; however, other complicated mechanisms are also involved. The Gigot and Schnelldorfer classifications are widely used in clinical practice. Most patients with PLD are asymptomatic; however, a few patients with advanced-stage disease may develop symptoms and complications that impair their quality of life and require treatment. The known treatment options for PLD are somatostatin analogues, aspiration with sclerotherapy, fenestration, hepatic resection, and liver transplantation. Although liver transplantation remains the only curative treatment for PLD, medical therapies are gradually being developed with the increasing knowledge of the disease’s pathophysiology. This review focuses on the clinical manifestations and diagnosis of PLD, as well as treatment strategies, to support clinicians regarding the clinical management of the disease.
Introduction
Polycystic liver disease (PLD) is a hereditary disease characterized by the presence of ≥20 liver cysts [1]. It presents in three forms: isolated autosomal dominant PLD (ADPLD), PLD with autosomal dominant polycystic kidney disease (ADPKD), and PLD with autosomal recessive polycystic kidney disease (ARPKD). However, the natural course and prognosis are similar among all the PLD classifications [1,2]. Genetic alterations, ciliary dysfunction of the biliary epithelial cells, and aberrant cell signaling pathways are main factors contributing to the complicated pathogenesis of PLD [3-6]. The severity of liver involvement strongly affects clinical symptoms, prognosis, and treatment strategies. Most patients with PLD are asymptomatic; however, patients in advanced stages may develop symptoms, including abdominal pain, gastrointestinal reflux, dyspnea, cystic infection and rupture, malnutrition, ascites, and variceal bleeding. These symptoms and complications seriously affect the quality of life [7,8]. Although liver transplantation is the only curative treatment for PLD, medical therapies are gradually being developed with the increasing knowledge of the disease’s pathophysiology [9-11]. This review focuses on the clinical manifestations, diagnosis, and treatment strategies for PLD to support clinicians in the clinical management of the disease.
Epidemiology and genetics
1. ADPLD
ADPLD is characterized by the presence of multiple liver cysts without renal involvement [1]. The incidence of ADPLD is reported in approximately 1/100,000 individuals worldwide [3]. Various genes are involved in the development of ADPLD; the most prevalent genes are PRKCSH and SEC63 which may account for 20% to 41% of the patients, followed by ALG8, LRP5, GANAB, and SEC61B (Table 1) [1,12-14]. Recently, PKHD1 gene mutation has been proposed to contribute to the development of ADPLD [15]. However, unlike the case with ADPKD and ARPKD, the mentioned genes account for only 30% to 50% of patients with ADPLD, and other pathogenic genes have not been identified in a large number of patients [13].

Major genes related to polycystic liver disease
2. PLD with ADPKD
ADPKD is the most frequent hereditary kidney disease, with a global incidence of 1/500 to 1/1,000 individuals [3]. PLD is the most common extra-renal manifestation of ADPKD [16]. The PKD1 and PKD2 genes have been reported as contributors in the development of ADPKD; PKD1 is reported to be associated with approximately 80% of patients with ADPKD, and PKD2 is reported to be associated with approximately 5% to 10% of patients [4,17]. Moreover, GANAB has recently been shown to be associated with ADPKD [17,18].
Pathophysiology
Genetic alterations, ciliary dysfunction of the biliary epithelial cells, and aberrant cell signaling pathways are crucial components in the mechanism of cystic development [4-6,20,21]. Although various genetic alterations contribute to the development of PLD, the exact mechanism is still unclear. To date, the most reliable hypothesis has been the two-hit theory. In addition to the occurrence of a germline mutation (first hit), a somatic mutation (second hit) is required for hepatic cystogenesis [22].
Most proteins involved in cystogenesis are located in the endoplasmic reticulum (ER). Mutations in the PRKCSH, SEC63, ALG8, LRP5, GANAB, and SEC61B genes are associated with the ER protein glycosylation [12,23]. PRKCSH encodes beta-subunit of glucosidase II and ALG8 encodes a glycosyltransferase, respectively. LRP5 is associated with signaling pathway including Wnt in the cyst development. GANAB encodes the catalytic alpha-subunit of glucosidase II which interacts with the beta-subunit of glucosidase II [24]. In addition, PKD1 and PKD2 genes in ADPKD encode polycystin-1 (PC-1) which is a ciliary protein necessary for cystogenesis, and polycystin-2 (PC-2) which modulates the intracellular calcium levels, respectively. PC-1 and PC-2 form a complex, which works on the surface of the ciliary membrane to simulate calcium uptake [25,26]. Dysfunction of ER proteins inhibits the synthesis, translocation, and expression of other proteins, including PC-1 [25,27]. Therefore, the deficiency in PC-1 expression and the dysfunction of PC-1/PC-2 complexes decrease the intracellular calcium levels and increase the cyclic adenosine monophosphate (cAMP) levels, thus contributing to biliary cell proliferation, fluid secretion, and cystic development [12,21,28].
Clinical presentation
Most patients with PLD show no clinical symptoms; however, a few patients develop symptoms that decrease their quality of life. The size, number, and location of cysts and the volume of the liver contribute to the development of a range of symptoms, including abdominal pain, abdominal distension, dyspnea, early satiety, gastroesophageal reflux, malnutrition, and back or flank pain caused by hepatomegaly [7-9,29]. The compression of the hepatic veins, portal veins, or inferior vena cava by hepatic cysts can result in a hepatic venous outflow obstruction and portal hypertension, with symptoms such as ascites, variceal bleeding, and splenomegaly [30,31]. Surprisingly, liver failure caused by PLD has been rarely reported; however, it may occur in very severe disease stages [7,14].
Risk factors associated with the progression of PLD include female sex, older age, multiple pregnancies, and prolonged estrogen exposure [32,33]. Moreover, a cohort study with a large sample size revealed that these factors contribute to symptom development in patients with PLD with ADPKD [14]. Another study revealed that most patients with PLD who underwent liver transplantation were females, suggesting that the disease advances more rapidly in females [34]. The number and size of hepatic cysts in patients with ADPKD are significantly correlated with the number of pregnancies, and some studies have reported an association between estrogen levels and liver volume in patients with PLD [32,33]. This may be supported by the fact that liver volume increases during the reproductive years and then stabilizes after menopause due to decreasing endogenous estrogen [9].
Complications
1. Hepatic cyst infection (HCI)
HCI is an uncommon complication with an incidence of approximately 1% of patients with hepatic cysts [35]. However, it is an important manifestation of PLD because it may lead to sepsis and death if left untreated. HCI is believed to occur because of the translocation of intestinal bacteremia. Its symptoms include upper right quadrant pain, malaise, and fever [1,36]. Imaging modalities such as computed tomography (CT) scans reveal cystic wall thickening with or without cell debris; however, these modalities are unreliable [36,37]. Recent studies have reported that 18-fluorodeoxyglucose positron emission tomography can be used to confirm HCI by detecting an 18-fluorodeoxyglucose accumulation in the infected cystic epithelia [38]. The gold standard for diagnosing HCI is the identification of inflammatory cells and bacteria via cystic aspiration [1,36,37]. Escherichia coli and Klebsiella spp. are the most prevalent bacteria detected through cystic aspiration [39]. Clinical, biochemical, and radiological parameters can be assessed if cystic aspiration is not possible. Broad-spectrum antibiotic therapy is recommended as the first-line treatment; however, patients with HCI who received only this therapy failed to achieve complete remission. Therefore, cystic drainage combined with antibiotic therapy should be considered [37].
2. Hepatic cyst hemorrhage
Hepatic cyst hemorrhage mainly occurs in large cysts and causes sudden pain in the upper right quadrant or flank [40]. High intra-cystic pressure, rapid cystic growth, and direct trauma are considered triggering factors for cystic hemorrhage. Cystic hemorrhage is diagnosed based on elevated Hounsfield units (due to fibrin aggregation) detected on imaging scans [41,42]. Conservative therapy is recommended in patients with minor symptoms; however, fenestration or hemorrhagic cyst removal can be considered in patients with severe symptoms [40].
3. Hepatic cyst rupture
Cystic rupture is a rare complication of PLD that occurs because of a significant increase in the cystic volume, either spontaneously or after the occurrence of cystic hemorrhage. An increased cystic size may cause instability and increase the risk of rupture. Severe acute abdominal pain is a typical symptom of hepatic cyst rupture. Imaging scans have revealed free fluid around a liver with a residual hepatic cyst. Conservative therapy is recommended in most cases. However, if hemodynamic instability or percutaneous ascites are detected, hepatic cyst drainage or surgical intervention may be required [43,44].
4. Portal hypertension
Patients with severe PLD may experience symptoms such as ascites, variceal hemorrhage, and splenomegaly due to portal hypertension [30]. Despite insufficient data on incidence of portal hypertension in patients with PLD, a retrospective study found that 6% of patients developed portal hypertension during the follow-up period [14]. Portal hypertension due to PLD can be classified into three types: hepatic venous outflow obstruction, portal vein obstruction and/or inferior caval vein syndrome. Most prevalent type is hepatic venous outflow obstruction [30]. Diagnosis is made based on imaging modalities including ultrasound, CT, and magnetic resonance imaging (MRI) by confirming hepatic cysts and ascites [45]. The detection of hepatic venous pressure gradient is needed for diagnosis basically based on the definition of portal hypertension, but it is rarely performed in patients with PLD because it is difficult to conduct due to complicated anatomy and it is usually applied as an assessment tool in patients with cirrhosis. The management of portal hypertension in patients with PLD is still challenging. Diuretics and repeated paracentesis are commonly used for management of ascites. Although there is a lack of data on the effect of somatostatin analogues on ascites control, it can be also used to reduce liver volume. In addition, surgical and interventional approaches for reducing liver volume, transjugular intrahepatic portosystemic shunts, hepatic or portal vein stenting can be considered. Liver transplantation should be considered in patients with refractory ascites or variceal bleeding that is not managed by conventional treatment [30].
Diagnosis
PLD is usually diagnosed based on the presence of ≥20 hepatic cysts [1]. Recently, the European Association for the Study of the Liver (EASL) has defined PLD as the presence of ≥10 hepatic parenchymal cysts not connected to the bile duct system [9]. A family history of ADPLD, ADPKD, or ARPKD as well as genetic tests can help in establishing the diagnosis. However, only 30% to 50% of patients with ADPLD showed genetic mutations, so genetic tests are not routinely performed to diagnose PLD [9,13,18].
Liver function tests are usually preserved in for most patients with PLD, because their liver parenchyma is not significantly damaged. Moreover, the carbohydrate antigen 19-9 levels may increase when hepatic cysts occupy an extensive part of the liver parenchyma or when an HCI develops [10,11]. Imaging scans are essential to identify hepatic cysts and exclude other diseases; ultrasonography (USG) and CT are the most widely used diagnostic methods because of their image quality, accessibility, and cost-effectiveness [46]. However, MRI is not routinely performed for diagnosis although it has a high sensitivity for detecting hepatic cysts and can be used to decide suitable treatment options [2,9].
Currently, three clinical classifications of PLD have been suggested: the Gigot [47], Schnelldorfer[48], and Qian classifications [7]. The Gigot and Schnelldorfer classification systems are widely used in clinical practice. According to the Gigot classification, PLD is classified into three types based on the number and size of hepatic cysts as well as the proportion of the remaining non-cystic liver parenchyma (Table 2). This classification focuses on performing hepatic cyst fenestration in suitable patients with PLD [47]. The Schnelldorfer classification classifies PLD into four types based on symptoms, cyst characteristics, areas of normal liver parenchyma, and portal or hepatic vein occlusion in the preserved sector (Table 3). This classification can be used to decide the optimal treatment [48]. The Qian classification is based on the number of cysts and presence of symptomatic hepatomegaly (Table 4) [7]. However, it is rarely used in clinical practice as it is insufficient for deciding the suitable treatment.

Gigot classification
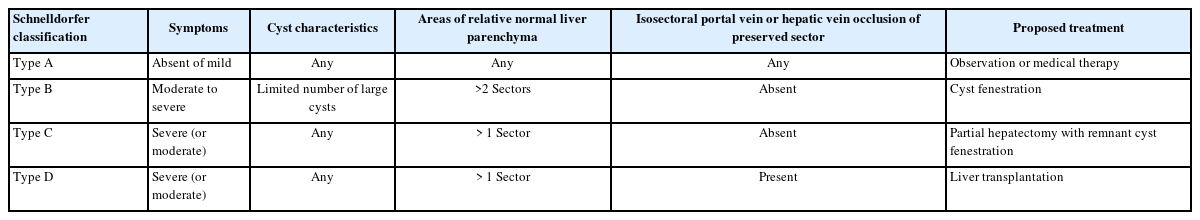
Schnelldorfer classification

Qian classification
Treatment
Treatment is not required for most asymptomatic patients with PLD; however, it is necessary for the symptomatic minority who have a low quality of life owing to the increased liver volume or complications resulting from PLD [29,49]. The primary goal of PLD treatment is to relieve symptoms and improve the quality of life [9,49]. Several studies have shown that estrogen contributes to the progression of PLD. Consequently, the EASL guidelines discourage female patients with PLD from using oral contraceptives [9].
PLD treatments are classified into three groups: medical, interventional, and surgical. The algorithm for the treatment of PLD is described (Fig. 1) [10]. The optimal treatment is decided according to the number, size, and location of hepatic cysts [9,10].
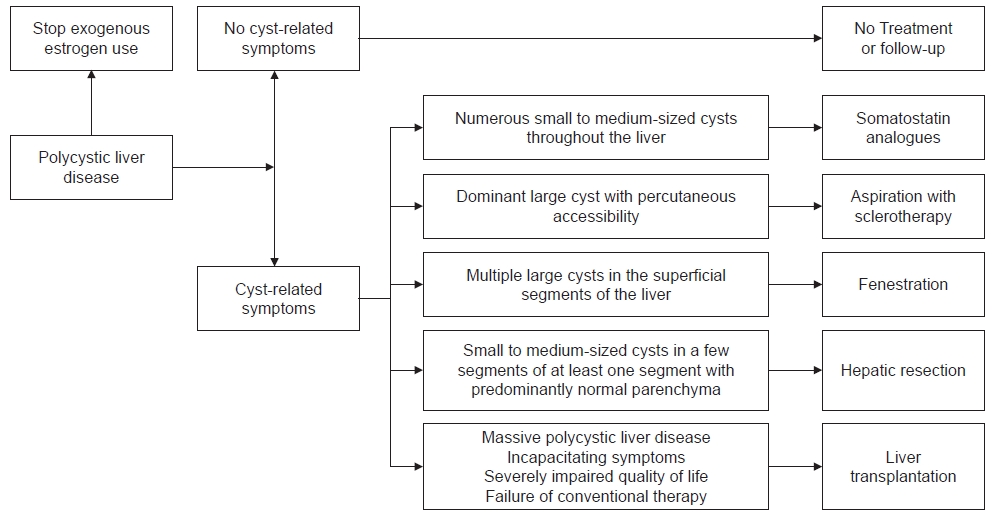
Treatment algorithm for patients with polycystic liver disease.
1. Medical treatment
1) Somatostatin analogues
Somatostatin is a cyclic peptide produced in various tissues, including the gastrointestinal tract and pancreas tissues. It regulates a wide range of physiological functions and hormones by binding to the somatostatin receptors (SSTR), which are classified into five subtypes: SSTR-1 to SSTR-5. The binding of somatostatin analogues to SSTR inhibits cAMP release in cystic cholangiocytes, thus decreasing cystic fluid production and inhibiting bile duct cell hyperplasia, leading to the prevention of hepatic cyst proliferation [50,51].
Multiple clinical trials have revealed that somatostatin analogues, such as octreotide, lanreotide, and pasireotide, prevent the progression of PLD by decreasing liver volume. Regarding octreotide, Pisani et al. [52] found that the total liver volume was significantly decreased by 130.2±133.2 mL (7.8%±7.4%) in patients who received octreotide per month; however, it was increased by 144.3±316.8 mL (6.1%±14.1%) in patients who received placebo after 3 years of treatment (p=0.004). The total liver volume reduction lasted for 2 years after the treatment completion. Moreover, Hogan et al. [53] reported that the total liver volume was significantly decreased by 4.95%±6.77% in the octreotide group (every 28 days) compared with that in the placebo group after 1 year of treatment (p=0.048). Regarding lanreotide, van Keimpema et al. [54] revealed that the mean liver volume was decreased by 2.9% in patients who received lanreotide per month; however, it was increased by 1.6% in those who received placebo after 6 months of treatment (p=0.01). Pasireotide, a somatostatin analogue with a long half-life, is used to treat Cushing’s syndrome [55]. An animal study revealed that pasireotide is more effective than octreotide in reducing hepatorenal cystogenesis in a rodent model [56]. Similarly, an in vivo study on pasireotide revealed that the total liver volume was changed by –3%±7% in the pasireotide group (every 28 days) compared with the 6%±7% increase in the placebo group. Therefore, researchers concluded that pasireotide slows the progressive-liver-volume increase [57].
Several meta-analyses have confirmed that somatostatin analogues lower liver volume and improve the quality of life [58-60]. Based on these findings, the EASL guidelines recommend administering somatostatin analogues for patients with numerous small to medium-sized hepatic cysts. The dosage of somatostatin analogues is needed to be adjusted based on efficacy and side effects [9]. In addition, somatostatin analogues are the most effective in young women whose hepatic cysts grow rapidly [61]. Somatostatin analogues are generally considered safe, and the occurrence of serious adverse effects resulting due to cessation is uncommon. Some patients may develop gastrointestinal symptoms (i.e., abdominal discomfort and diarrhea) as well as gallbladder stones [62,63]. Further studies are required to evaluate the effects of long-term maintenance therapy because most studies on somatostatin analogues are based on relatively short treatment periods ranging from 6 months to 3 years.
2) Mechanistic target of rapamycin (mTOR) inhibitors
mTOR inhibitors are a class of drugs that inhibit mTOR, a serine/threonine-specific protein kinase that belongs to the phosphatidylinositol-3 kinase related kinases family. mTOR regulates various cellular metabolic pathways by signaling through two protein complexes, mTORC1 and mTORC2 [64]. Currently, mTOR inhibitors, including sirolimus and everolimus, are used to treat cancer [65]. In some animal studies on PKD, mTOR inhibitors have been reported to be effective in preventing the growth of hepatic cysts [66]. However, only a few clinical trials on mTOR inhibitors have been conducted, and these mentioned findings have not been confirmed in the clinical trials. Chrispijn et al. [67] evaluated the effectiveness of everolimus plus octreotide versus octreotide monotherapy in reducing liver volume in patients with PLD. No statistically significant differences were detected between the two groups (p=0.73). Furthermore, mTOR inhibitors cause many adverse events including interstitial lung disease, thrombosis, rash, anemia, hyperglycemia, dyslipidemia, and an increased risk of infection [68]. Therefore, mTOR inhibitors are not recommended for the treatment of PLD because of their toxicity and because of insufficient evidence [9].
3) Vasopressin-2 receptor (V2R) antagonists
V2R is predominantly located in the distal tubules and collecting ducts of the kidney, and its activation induces an elevation in the level of cAMP, which stimulates cell proliferation and hepatic cyst growth [69]. Although V2R antagonists have been clinically confirmed for the treatment of ADPKD [70,71], very few studies have investigated their effectiveness in treating PLD. Only a few case reports have shown that tolvaptan has a favorable effect on reducing liver volume in patients with PLD with ADPKD [72,73]. Therefore, further studies are required to assess the efficacy of V2R antagonists in managing PLD.
4) Ursodeoxycholic acid
Ursodeoxycholic acid (UDCA), which is widely administered in patients with chronic liver disease, increases intracellular calcium levels in hepatocytes and biliary epithelial cells [74]. Based on this mechanism, it had a favorable effect on delaying the development of hepatic cysts in a rat model. However, only few studies on the efficacy of UDCA in patients with PLD have been conducted. In a phase 2 multicenter randomized controlled study including 34 patients with PLD, total liver volume increased by 4.6%±7.7% after 24 weeks of UDCA treatment compared to 3.1%±3.8% in the control group (p=0.493) [75]. UDCA is not currently recommended for the treatment of PLD according to EASL guidelines [9].
2. Interventional treatment
1) Cystic aspiration with sclerotherapy
Cystic aspiration with sclerotherapy is commonly performed for symptomatic patients with dominant large cysts classified as Gigot type I or Schnelldorfer type B to reduce liver volume [1,4,76]. This procedure is conducted under imaging guidance such as USG. After the aspiration of cystic fluid, a sclerosing agent is injected. Ethanol is the most commonly used sclerosing agent, followed by ethanolamine oleate, minocycline, and tetracycline [76,77]. Sclerosing agents destroy the epithelial lining of the cystic wall, thus preventing fluid accumulation within the cyst [78]. Although a single procedure is typically sufficient to treat dominant cysts, some patients require a series of procedures for cystic elimination or to achieve symptomatic relief [79].
In a systematic analysis of 16 studies assessing the efficacy of aspiration with sclerotherapy for hepatic cysts, including 526 patients with a total of 588 cysts, 76% to 100% of patients reported a partial reduction in the cystic volume, and 72% to 100% reported symptom improvement [80]. Its side effects include postprocedural pain caused by the sclerosing agent and hepatic cyst hemorrhage; however, no fatal complications have been reported [80]. Despite its efficacy for hepatic cysts, aspiration with sclerotherapy is rarely performed in clinical practice because most patients with PLD have multiple hepatic cysts and/or because the cystic size is not suitable for performing the procedure [11].
2) Transcatheter arterial embolization (TAE)
TAE for patients with PLD is based on the concept that the cysts are mostly supplied by the hepatic arteries. Impaired blood supply induces the destruction of the cystic epithelial cells, thus reducing cystic fluid accumulation and inhibiting disease progression [81,82]. In a retrospective study on 244 patients with PLD, who underwent TAE, liver volume decreased by 94.7% of the pre-treatment volume within 6 months and by 90.8% within 12 months [83]. However, in a study on 18 patients who underwent TAE, 69.6% experienced TAE failure which included the occurrence of refractory symptoms, cystic infection, liver failure, and death [84]. Therefore, further controlled studies are required to evaluate the efficacy and safety of TAE and to clinically examine this procedure in patients with PLD.
3. Surgical treatment
1) Fenestration
Cystic fenestration is a surgical procedure that combines aspiration and deroofing of hepatic cysts [85]. Fenestration is usually indicated for patients with Gigot type I-II or Schnelldorfer type B PLD, especially those with cysts in the superficial segments of the liver [6,11]. Additionally, it can be considered when cystic aspiration with sclerotherapy fails. The main advantage of fenestration is that multiple cysts can be treated in a single session [86,87]. Its common complications include ascites, hemorrhage, pleural effusion, and bile leakage [85]. Currently, laparoscopic fenestration is preferred to open fenestration as it has the advantages of shorter hospital-stay duration, fewer complications, and the absence of significant differences regarding the outcomes. Nevertheless, open fenestration is appropriate in cysts located in difficult-to-access regions such as the hepatic dome or right posterior segments [85,87]. In a meta-analysis that assessed symptomatic relief and recurrence after laparoscopic fenestration in 1,314 patients with hepatic cysts, 90.2% of the patients showed symptomatic relief. The rates of symptomatic recurrence and re-intervention were 9.6% and 7.1%, respectively. However, in a subgroup analysis on patients with PLD (n=146), the rates of symptomatic recurrence, re-intervention, and complications were significantly higher (33.7%, 26.4%, and 29.3%, respectively) [88]. Cystic fenestration is considered a suitable treatment option in patients with symptomatic large cysts.
2) Hepatic resection
Hepatic resection is primarily performed in symptomatic Gigot type II or Schnelldorfer type C PLD with small to medium-sized cysts in a few segments with at least one segment having predominantly normal liver parenchyma [11,48]. In addition, it can be considered when aspiration with sclerotherapy, cystic fenestration, or liver transplantation is not available [6,10]. Hepatic resection results in a significant reduction in liver volume and symptomatic relief. However, the biliary tract, vascular structure, and Glisson’s capsule distortions caused by cystic compression and hepatic venous outflow obstruction complicate the operation and increase morbidity and mortality rates. The main complications include intra- or post-operative hemorrhage, ascites, pleural effusion, and liver failure [4,86]. A review of 26 articles on 337 patients with PLD who underwent hepatectomy revealed that 86% of the patients showed symptomatic relief. The morbidity and mortality rates were 51% and 3%, respectively. Cystic recurrence was noted in 34% of the patients [86]. Currently, studies on combining partial hepatectomy with cystic fenestration [85] and somatostatin analogues administration after hepatectomy in patients with PLD [89] have shown favorable outcomes in terms of liver volume reduction and hepatic cyst growth suppression. Although hepatic resection relieves symptoms and reduces liver volume, we should keep in mind its considerable morbidity and mortality rates, as well as the potential challenges we may encounter when performing future liver transplantation due to the occurrence of adhesions [4,10,86,90].
3) Liver transplantation
Liver transplantation is the only curative treatment for PLD and is mostly performed in patients with Gigot type III or Schnelldorfer type D disease [4,9,10,48]. Patients with incapacitating symptoms such as severe malnutrition, portal hypertension, ascites, variceal bleeding, or recurrent HCIs or those who have failed conventional therapy should be considered for undergoing liver transplantation [91,92]. According to the European Liver Transplant Registry study, the 1- and 5-year graft survival rates were 94.3% and 87.5%, and the 1- and 5-year survival rates were 94.8% and 92.3%, respectively [34].
The Model for End-Stage Liver Disease (MELD) score, which has been validated in patients with liver cirrhosis, is currently the main criteria for deciding to allocate liver grafts [93,94]. However, it is difficult to allocate liver grafts in patients with PLD because of the low MELD scores, as liver function remains preserved even in the most advanced stage. As a result, several exceptions have been proposed to take patients with PLD on waiting lists into consideration. support patients with PLD on waiting lists [91,95]. These guidelines need to be applied more effectively in the allocation system, and more beneficial criteria are required.
Combined liver-kidney transplantation should be considered in patients with PLD with ADPKD and severe renal impairment (creatinine clearance <30 mL/min) since the outcome of this combined technique has been better than that of liver transplantation alone [96,97]. However, combined liver-kidney transplantation have been raised rates of short-term kidney graft loss about 20% [98]. Therefore, a multidisciplinary approach for decision-making in selecting patients, involving hepatologists and nephrologists, is recommended.
Conclusion
PLD is an inherited genetic disorder. Only a few patients develop symptoms and complications that require further treatment. Its diagnosis is based on a family history of the disease and the presence of multiple hepatic cysts, with or without renal cysts, via imaging modalities. The primary goal of PLD treatment is to relieve symptoms and improve the patients’ quality of life. Liver transplantation is the only curative treatment option; however, it is not available for many patients. In addition to liver transplantation, medical therapies including somatostatin analogues, aspiration with sclerotherapy, TAE, fenestration, and liver resection can be carefully applied in certain situations. Future effective pharmacological treatments or combination therapies may be developed based on a better understanding of the PLD pathophysiology.
Notes
Conflicts of interest
Hyun Joon Park is an editorial board member of the journal but was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Funding
None.
Author contributions
Conceptualization: HJP. Supervision: HJP. Writing – original draft: JJ. Writing – review & editing: HJP.